Enfermedades Metabólicas Hereditarias: clasificación basada en la fisiopatología
Referencia: A. García-Cazorla, J.M. Saudubray. En: Diagnóstico y tratamiento de las enfermedades metabólicas hereditarias 5ª edición. Eds. Mª Luz Couce, Luis Aldámiz-Echevarría, Mª Concepción García-Jiménez, Domingo González-Lamuño. Ergon (Madrid) 2022, pags: 25-31.
1.- ¿Qué son las Enfermedades Metabólicas Hereditarias o Errores Innatos del Metabolismo?
El metabolismo es una enorme red de reacciones químicas de gran complejidad que modula las funciones celulares y comprende miles de moléculas, fundamentalmente proteínas, enzimas, cofactores, receptores y transportadores. Las Enfermedades Metabólicas Hereditarias (EMH), también denominados Errores innatos del Metabolismo (EIM) son defectos de origen genético que ocasionan disfunciones en algún elemento de esta intrincada red. En los últimos años, las técnicas genéticas de secuenciación masiva han permitido describir una gran cantidad de nuevos EIM. Muchos de ellos no presentan biomarcadores detectables con las técnicas bioquímicas estandarizadas y utilizadas en la mayoría de centros de referencia para estas enfermedades, lo cual no excluye que otras técnicas (como las -ómicas) puedan determinarlos en el futuro. Estos avances han llevado a la comunidad científica a reflexionar sobre el concepto mismo de EIM, introducido por Garrod al describir las primeras enfermedades hereditarias del metabolismo en 1908.
Según Morava, para que un defecto genético sea considerado como EIM solo se requiere que la afectación de enzimas específicas o vías bioquímicas sea intrínseca al mecanismo fisiopatológico (1). De este modo, y considerando esta definición, si hace alrededor de una década se estimaban unos 600 EIM, en la actualidad se reconocen algo más de 1.400 EIM según IEMbase ( http://www.iembase.org).
Este crecimiento tan importante en el número de enfermedades ha traído consigo la definición de nuevas categorías fisiopatológicas como los defectos de la síntesis y remodelación de los lípidos complejos (2) o defectos de la autofagia (que a su vez pertenecen a la gran categoría de defectos del tráfico celular) (3). Atendiendo a la necesidad de reunir a los nuevos EIM bajo un mismo paraguas y de un modo estructurado, en los últimos dos años han aparecido diversas iniciativas de nosologías y clasificaciones que describimos brevemente a continuación:
- A principios de 2019 Ferreira y cols. proponen una nosología de EIM que incluye en el momento de su publicación alrededor de 1.100 EIM distribuidos en 130 grupos, y que establece una serie de criterios de definición de EIM. Entre ellos destaca “la disrupción de una vía metabólica independientemente de las alteraciones de laboratorio” (4).
- Unos meses más tarde, en julio de 2019, Saudubray y cols. (5) proponen una clasificación simplificada basada en la fisiopatología y en la presentación clínica, que considera e integra todos los EIM incluidos en la nosología previa.
- Tras el esfuerzo conjunto de múltiples expertos y sociedades científicas, se ha propuesto una clasificación internacional de los EIM (ICIMD) que incluye 1.442 enfermedades divididas en 23 grupos y que ha sido publicada muy recientemente (6).
Dada la complejidad del tema, basaremos este capítulo en la clasificación simplificada, que además establece vínculos directos con la presentación clínica (Figs. 1, 2 y 3).
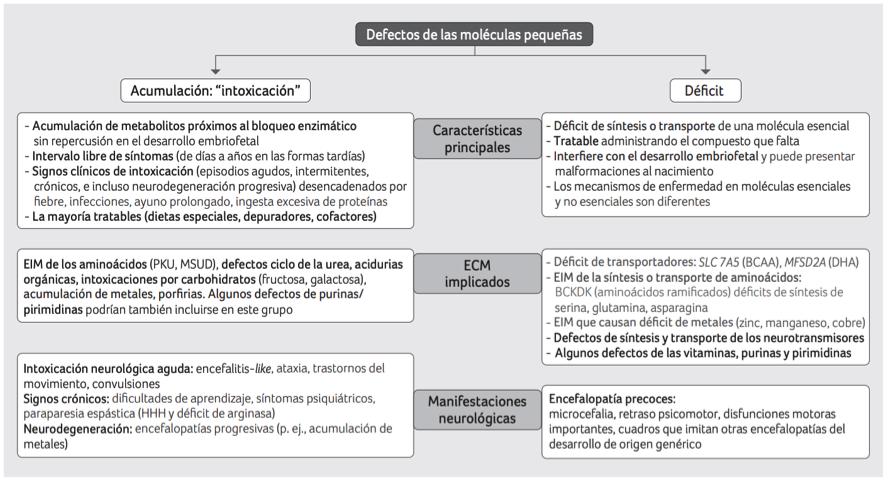
FIGURA 1.
EIM de las moléculas pequeñas. Las señaladas en gris corresponden a las enfermedades descritas más recientemente. (Adaptada de: García-Cazorla A, Saudubray JM. New insights in inborn errors of metabolism are leading to new paradigms in child neurology. Rev Neurol. 2018; 66 [Suppl 2]: S37-42).

FIGURA 2.
EIM de las moléculas complejas. Las señaladas en gris corresponden a las enfermedades descritas más recientemente. (Adaptada de: García-Cazorla A, Saudubray JM. New insights in inborn errors of metabolism are leading to new paradigms in child neurology. Rev Neurol. 2018; 66 [Suppl 2]: S37-42).

FIGURA 3.
EIM relacionadas con el metabolismo energético. Las señaladas en gris corresponden a las enfermedades descritas más recientemente. (Adaptada de: García-Cazorla A, Saudubray JM. New insights in inborn errors of metabolism are leading to new paradigms in child neurology. Rev Neurol. 2018; 66 [Suppl 2]: S37-42).
2.- Clasificación simplificada en la fisiopatología de los EIM.
Esta clasificación simplificada se basa en los siguientes principios:
- El razonamiento médico en la práctica diaria necesita partir de las manifestaciones clínicas. El paciente acude a la consulta con una serie de síntomas que se intentan relacionar con los grupos o categorías principales de EIM susceptibles de ocasionar esa clínica en particular. Por lo tanto, una clasificación que incluya fenotipos clínicos y mecanismos de enfermedad agrupados en pocas categorías es útil en la práctica clínica no solo para médicos especialistas en medicina metabólica, sino también para pediatras generales, neurólogos, internistas, endocrinólogos y otros.
- La propuesta de una clasificación simplificada se centra también en tratamientos basados en fisiopatología. Por lo tanto, estimula el ejercicio clínico de vincular síntomas, fisiopatología y manejo terapéutico, particularmente en situaciones de emergencia.
- Las descripciones exhaustivas y precisas de cada enfermedad son esenciales para ampliar y desgranar el gran universo de las patologías metabólicas. Sin embargo, un enfoque holístico e integrador es crucial para no perder de vista las bases comunes de esta compleja especialidad.
- La combinación de la bioquímica con procesos de biología celular es actualmente imprescindible para abordar el paisaje fisiopatológico de los EIM.
- La propuesta de una clasificación simplificada de los EIM se basa en tres grandes categorías fundamentadas en el tamaño de las moléculas (“pequeñas” o “complejas”) y su implicación en el metabolismo energético. Incluye conceptos y patologías clásicas, descritas en fases muy tempranas del desarrollo de la medicina metabólica, pero también aspectos totalmente innovadores.
3.- GRUPO 1: EIM DE LAS MOLÉCULAS PEQUEÑAS
Casi todos estos EIM tienen marcadores metabólicos en plasma y/o orina que se pueden medir fácil y rápidamente en caso de emergencia, generalmente en una sola determinación. Estos marcadores incluyen aminoácidos (AA), ácidos orgánicos, acilcarnitinas, porfirinas, ácidos grasos (AG), purinas, pirimidinas, metales o metabolitos de galactosa. De igual modo pueden utilizarse para monitorizar los tratamientos, ya que la mayoría de estos IMD son susceptibles a algún tipo de abordaje terapéutico. Este grupo se puede dividir en dos subcategorías (Fig. 1).
3.1. Acumulación de moléculas pequeñas: enfermedades por “intoxicación”
La acumulación de moléculas pequeñas causa trastornos agudos o progresivos por “intoxicación”. Este grupo de EIM corresponde a enfermedades clásicas, descritas desde los inicios de la medicina metabólica. Los signos y síntomas resultan principalmente de la acumulación anormal de los compuestos proximales al bloque. No interfieren con el desarrollo neurológico embrionario y fetal y se presentan después de un intervalo sin síntomas (días a años) con signos clínicos de “intoxicación” (aguda, intermitente, crónica e incluso progresiva que conduce a la neurodegeneración) provocada por el ayuno, el catabolismo, la fiebre, enfermedades intercurrentes e ingesta de alimentos. La mayoría de estos trastornos son tratables y requieren la eliminación de la “toxina” por dietas especiales, depuradores y cofactores (principalmente vitaminas).
Este grupo comprende EIM del catabolismo de aminoácidos, como la fenilcetonuria, la leucinosis, los defectos del ciclo de la urea, las acidurias orgánicas, la galactosemia y enfermedades por acumulación de metales, como la enfermedad de Wilson, las neuroferritinopatías y los síndromes de acumulación de hierro cerebral, así como la hipermanganesemia (SLC30A10 y SLC39A14) (7). La acumulación de algunas moléculas pequeñas puede dar lugar a cristales visibles en diversos tejidos, como la cistina en la cistinosis (un defecto lisosomal), el oxalato en la oxalosis (un defecto peroxisomal) o el ácido homogentisico (ocronosis) en la alcaptonuria. Algunos defectos de las purinas y las pirimidinas también pueden clasificarse en este grupo.
Los defectos de reparación de metabolitos son una nueva categoría de EIM en la que los síntomas están relacionados con la acumulación de compuestos tóxicos como en la aciduria L-2-hidroxiglutárica, o mutaciones NAXE: NAXE cataliza la epimerización de NAD (P) HX, evitando así la acumulación de metabolitos tóxicos (8).
Las vitaminas (transporte y procesamiento intracelular) interfieren con muchas vías metabólicas diferentes donde actúan como cofactores enzimáticos, chaperonas o moléculas de señalización.
En la mayoría de los trastornos que responden a las vitaminas, los niveles plasmáticos de vitaminas son normales y el tratamiento se basa en dosis farmacológicas de suplementos vitamínicos (dependencia de vitaminas). Por el contrario, algunos otros defectos sensibles a las vitaminas relacionados con la absorción o el transporte de la vitamina o su precursor, como la deficiencia secundaria de niacina debido a la malabsorción de triptófano en la enfermedad de Hartnup, la deficiencia de folato en los defectos de FOLR o la tiamina y los defectos de transporte de riboflavina, se caracterizan por bajos niveles de vitamina en plasma (o LCR) y responden a dosis fisiológicas de vitamina (deficiencia de vitaminas). Sin embargo, la mayoría de estos IMD que afectan el metabolismo de las vitaminas pueden convertirse en trastornos de intoxicación. Por ejemplo, tanto la mala absorción de vitamina B12 como los defectos intracelulares de cobalamina conducen a la acumulación de homocisteína y ácido metilmalónico (MMA).
Las hiperglicinemias y el defecto del transportador de dopamina (acumulación de ácido homovanílico en la hendidura sináptica) se comportan más como trastornos de los neurotransmisores y moléculas de señalización que como una intoxicación. De hecho, en el cerebro, las moléculas que se acumulan actúan como moléculas de señalización que activan vías biológicas relacionadas con la plasticidad neuronal, la excitabilidad y la supervivencia neuronal.
3.2. Deficiencia de moléculas pequeñas
Los síntomas resultan principalmente de la síntesis defectuosa de compuestos que son distales al bloqueo o del transporte defectuoso de una molécula esencial a través de las membranas celulares u organelas. Los signos clínicos son, al menos en teoría, tratables al proporcionar el compuesto faltante. La mayoría de estos defectos afectan el desarrollo neurológico y pueden tener una presentación congénita (prenatal). Comparten características con los trastornos del grupo de moléculas complejas (véase más adelante).
Este grupo abarca todos los defectos de moléculas esenciales que deben transportarse a través de las membranas celulares, errores innatos de la síntesis de AA y FA no esenciales y varios trastornos de las pirimidinas que afectan la síntesis de nucleósidos de citidina, uridina y timidina. Son potencialmente tratables, como la deficiencia de CAD (Koch y cols. 2017) o la aciduria orótica congénita. Los EIM más paradigmáticos vinculado a defectos de transporte son el transporte defectuoso de los AA ramificados (BCAA) (9), debido a mutaciones en el gen SLC7A5, y la deficiencia de MFSD2A que resulta en el transporte defectuoso de un AG esencial, el ácido docosahexanoico (DHA) (10). La deficiencia de la quinasa de la deshidrogenasa sobreactiva la oxidación de BCAA y conduce a niveles muy bajos de BCAA. Se presenta como una enfermedad del neurodesarrollo con signos dentro del espectro del autismo y epilepsia, similar al defecto de transporte. Ambas producen una enfermedad del neurodesarrollo con signos dentro del espectro autista y epilepsia. Otros defectos de transporte de AA esenciales están vinculados a mutaciones en SLC7A7 y SLC6A19. Estos son responsables de la intolerancia a las proteínas dibásicas (lisina y aminoácidos dibásicos) y la enfermedad de Hartnup (aminoácidos neutros con deficiencia secundaria de niacina), respectivamente.
Ambas enfermedades muestran niveles bajos de aminoácidos en plasma y pueden presentarse con manifestaciones multisistémicas posnatales. Por el contrario, los defectos de transportador de AA no esenciales (SLC3A1, SLC7A9, SLC1A1) que involucran cistina, glicina, prolina, hidroxiprolina, ácidos dicarboxílicos se presentan solo con hiperaminoaciduria y permanecen principalmente asintomáticos o con síntomas “localizados” (urolitiasis).
Los defectos de síntesis de AA no esenciales también pueden presentarse prenatalmente con defectos del desarrollo neurológico incluyendo malformacions corticales extensas. Las formas graves de defectos en la síntesis de serina causan el síndrome de Neu Laxova (11).
Las deficiencias de glutamina sintetasa y asparagina sintetasa muestran una agiria casi completa y se manifiestan clínicamente como encefalopatías epilépticas congénitas graves.
Los AG, ya sean derivados de fuentes dietéticas o sintetizado de novo, se puede convertir en AG de cadena más larga para incorporarlos en lípidos complejos. Las deficiencias de elongasas ELOVL5 y ELOVL4 causan ataxia espinocerebelosa dominante en adultos, SCA38 y SCA34, respectivamente. El ELOVL4 autosómico recesivo y el déficit de ELOVL1 pueden presentarse en la infancia con disrupción del neurodesarrollo e ictiosis, similares al síndrome de Sjogren-Larsson. Los defectos de síntesis/elongación de AG comparten muchas similitudes con los trastornos catabólicos peroxisomales de ácidos grasos de cadena muy larga y las deficiencias de síntesis y remodelación de lípidos complejos. Además de los defectos de AA y AG, el grupo de defectos de molécula pequeña también abarca EIM que afectan a neurotransmisores y deficiencia de metales. El síndrome de deficiencia del transportador de manganeso, causado por mutaciones SLC39A8, muestra neurodegeneración severa de inicio tardío con bajo nivel de manganeso en plasma (12).
En resumen, la mayoría de los trastornos relacionados con deficiencias de moléculas pequeñas producen importantes alteraciones del desarrollo neurológico, lo que conduce a encefalopatías globales graves donde casi todas las funciones neurológicas se alteran crónicamente. En las presentaciones de inicio temprano, los pacientes muestran retrasos psicomotores severos que afectan los hitos motores y cognitivos. Estos defectos imitan a las encefalopatías genéticas “no metabólicas” tempranas que afectan las funciones cruciales del desarrollo neurológico. Curiosamente, las formas de inicio tardío se presentan como trastornos neurodegenerativos, lo cual refleja los diferentes papeles del metabolismo a medida que el cerebro envejece.
4. GRUPO 2: EIM DE LAS MOLÉCULAS COMPLEJAS
Este grupo en expansión abarca enfermedades que alteran el metabolismo de moléculas complejas que no son solubles en agua ni difusibles. Dichas moléculas complejas ubicuas son: glucógeno, esfingolípidos (SPL), fosfolípidos (PL), ácidos biliares, glucosaminoglucanos, oligosacáridos, glicoproteínas, glicolípidos y ácidos nucleicos. A estos se añaden AG y colesterol de cadena muy larga, aunque son moléculas simples, ya que pueden ser una fuente de moléculas complejas, como glicolípidos, PL, SPL y ésteres de colesterol, cuya deficiencia comparten muchas similitudes clínicas con los lípidos complejos.
Los procesos de síntesis y reciclaje de moléculas complejas tienen lugar en organelas (mitocondrias, lisosomas, peroxisomas, retículo endoplásmico, aparato de Golgi y vesículas sinápticas) y la mayoría de las vías involucran varios orgánulos y requieren transportadores de proteínas o vesículas. Los trastornos congénitos de la glicosilación y tráfico, procesamiento y control de calidad pertenecen a esta categoría. De hecho, los defectos de moléculas complejas ejemplifican el nuevo paradigma de conexiones entre las rutas metabólicas y las organelas subcelulares. Además, múltiples mecanismos fisiopatológicos pueden estar presentes dentro de una sola enfermedad. Como ejemplo, la mutación SLC10A7 es un trastorno de la N-glicosilación que da lugar a un defecto de biosíntesis de glicosaminoglucanos y una displasia esquelética (13). En este grupo, los síntomas clínicos son permanentes, a menudo progresivos e independientes de eventos intercurrentes, no relacionados con la ingesta de alimentos. La mayoría de las enfermedades no se presentan con crisis metabólicas.
De manera similar al grupo de moléculas pequeñas, también hay dos subcategorías de trastornos de moléculas complejas.
4.1. Acumulación de moléculas complejas
Los defectos del catabolismo conducen al almacenamiento de un compuesto en el citoplasma (glucogenosis, esteatosis) o en lisosomas. Son el grupo más típico e histórico (como las esfingolipidosis o las mucopolisacaridosis) en el que los signos y síntomas son principalmente el resultado de la acumulación anormal de uno o varios compuestos proximales al bloqueo. En general, no hay manifestación prenatal, aunque algunas formas graves se presentan como hidrops o malformaciones (14). Las presentaciones neurológicas muestran trastornos progresivos con neurodegeneración de inicio tardío con o sin signos obvios de “almacenamiento”. El diagnóstico se basa principalmente en la detección de metabolitos en orina (mucopolisacáridos, oligosacáridos, sulfatidas, ácido siálico) y análisis de enzimas leucocitarias además de los estudios genéticos. Un número creciente de defectos de la autofagia y el tráfico celular pueden imitar clínicamente las enfermedades lisosomales clásicas.
4.2. Deficiencia de moléculas complejas
4.2.1. Los déficits de glucógeno se han denominado también “glucogenosis” en este caso de tipo 0a y 0b (glucógeno sintasa hepática y muscular, respectivamente), vinculados a mutaciones GYG1/2 que codifican para glucogenina. Este grupo también está relacionado con el metabolismo energético.
4.2.2. Los defectos de síntesis y remodelación de fosfolípidos (PL), glicoesfingolípidos (GSL) y de cadena larga, muy larga, cadena ultra larga (AG) representan un nuevo grupo en rápida expansión (2).
Los defectos de síntesis y remodelación de PL y GSL conducen a una variedad de síntomas neurodegenerativos progresivos, miopatía y miocardiopatía (como en el síndrome de Barth o Sengers), signos ortopédicos (hueso y condrodisplasia, malformación), ictiosis sindrómica y distrofia retiniana. Los fosfatidilinositoles (P-ins) son PL regulados por P-ins 3-quinasas, enzimas de señalización que regulan una amplia gama de procesos, incluidos el crecimiento celular, la proliferación, la migración, el metabolismo y el desarrollo del cerebro. Muchas mutaciones que afectan a este sistema causan trastornos del neurodesarrollo y neurodegenerativos, como varias ataxias congénitas relacionadas con mutaciones en el receptor de inositol 1,4,5-trifosfato (IP3R) o síndromes de Joubert. En general, dada la vida media muy corta de los P-ins, no hay un marcador metabólico fácil para estas enfermedades y el diagnóstico se basa en herramientas de secuenciación masiva.
4.2.3. Los trastornos peroxisomales abarcan una serie de defectos que afectan la biogénesis del peroxisoma o enzimas específicas del catabolismo de ácidos grasos de cadena muy larga (AGCML) y de cadena ramificada (ácido fitánico) o síntesis de moléculas complejas como ácidos biliares o plasmógenos. Muchos se presentan en periodo neonatal como síndrome polimaformativo (p. ej., síndrome de Zellweger). Otros se presentan más tarde entre la primera y segunda década de la vida o en la edad adulta, en forma de trastornos neurodegenerativos (enfermedad de Refsum, X-ALD/AMN). La síntesis de plasmógenos (una subcategoría de PL) involucra no solo los peroxisomas, sino también el retículo endoplásmico (RE). Como ejemplo, la deficiencia de ACBD5 interrumpe la transferencia de lípidos entre el RE y los peroximas y produce una síntesis disminuida de plasmógenos asociados a una leucodistrofia progresiva con ataxia y distrofia retiniana (15). El diagnóstico se realiza mediante la determinación en sangre de AGCML, ácidos fitánico, pristánico y pipecólico en plasma y plasmógenos en eritrocitos.
4.2.4. Los defectos de síntesis de colesterol y ácidos biliares se presentan con síndromes polimalformativos, como el síndrome de Smith-Lemli-Opitz (SLO), colestasis neonatal o trastornos neurodegenerativos de inicio tardío, como la xantomatosis cerebrotendinosa tratable con ácido quenodesoxicólico. La fisiopatología de los trastornos de los esteroles es compleja y puede ser el resultado de la deficiencia de colesterol durante el desarrollo embrionario, la acumulación de intermediarios tóxicos de esteroles proximales a cada bloqueo enzimático, la generación de oxisteroles anormales y/o la señalización anormal por proteínas hedgehog que normalmente contienen uniones colesterol. El diagnóstico se realiza mediante el perfil de colesterol y oxiesteroles. La deficiencia de escualeno sintasa es un defecto de síntesis de colesterol recientemente descrito. Los pacientes se presentan con dismorfia facial (tipo SLO) y compromiso neurológico grave. Los niveles de colesterol en plasma son bajos y el perfil de ácidos orgánicos en orina es anómalo (16).
4.2.5. Defectos de síntesis de glicosaminoglicanos (GAG): los GAG se construyen mediante la adición gradual de monosacáridos por diversas glicosiltransferasas y están modulados por epimerasas y sulfotransferasas. Juegan una amplia gama de actividades biológicas.
Las mutaciones en los genes humanos que codifican glicosiltransferasas, sulfotransferasas y enzimas relacionadas son responsables de la biosíntesis de los GAG. Deben sospecharse en pacientes con una combinación de características clínicas características en tejido conectivo: hueso y cartílagos (huesos largos y cortos con o sin escoliosis), ligamentos (laxitud/dislocaciones articulares) y el subepitelio (piel, escleras). Algunos de estos defectos comparten características con los CDG. El análisis de GAG, los estudios enzimáticos y genéticos proveen el diagnóstico. Los trastornos de síntesis de oligosacáridos se clasifican como trastornos congénitos de glicosilación (véase más adelante).
4.2.6. Los trastornos de los ácidos nucleicos son mucho más que los defectos clásicos de purina y pirimidina si consideramos defectos de sintetasas de ARNt citoplasmático y mitocondrial, ribosomopatías(17), enfermedades que afectan los mecanismos de la reparación del ADN/ARN (diversos subtipos del síndrome de Aicardi-Goutières y los trastornos relacionados con la metilación del ADN [ADNm]), como los síndromes CHARGE y Kabuki, en los que se han identificado recientemente firmas de ADNm altamente específicas y sensibles.
4.3. Trastornos de tráfico y procesamiento celular
4.3.1. Defectos de la glicosilación (CDG): incluye cualquier condición clínica inexplicable, particularmente en la enfermedad multiorgánica con afectación. Muchos CDG interfieren con el desarrollo neurológico en la vida fetal, principalmente los trastornos de O-glicosilación y relacionados con el glicosilfosfatidilinositol. Los métodos de detección se limitan al análisis de transferrina sérica y apolipoproteína C-III sérica. La glicómica clínica podría aportar un diagnóstico muy eficiente. Los estudios genéticos son cada vez más útiles en el diagnóstico de pacientes con CDG-X. La clasificación detallada de este vasto y complejo grupo en crecimiento está más allá del alcance de esta revisión.
4.3.2. Defectos de vesiculación intracelular, tráfico, procesamiento de moléculas complejas y los procesos de control de calidad (como el plegamiento de proteínas y la autofagia): esto se ejemplifica en EIM com el síndrome neurocutáneo CEDNIK, debido a la mutación en la codificación SNAP 29 para una proteína SNARE implicada en la vesiculación intracelular, mutaciones en AP5Z1, la cual codifica una proteína implicada en el tráfico vesicular, que causa paraplejía espástica hereditaria, y cuyo fenotipo celular se asemeja notablemente a las enfermedades lisosomales por depósito. Mutaciones en CHMP2B y GRN, codifican la proteína del cuerpo multivesicular cargada y progranulina, respectivamente; y se caracterizan por una patología de almacenamiento lisosomal neuronal que se presenta con demencia frontotemporal familiar. De manera similar, mutaciones en ZFYVE20 codifican para Rabenosyn-5 (afectación de la endocitosis) con un fenotipo complejo que incluye convulsiones intratables. Los defectos de la autofagia son EIM del tráfico celular que se presentan como trastornos neurometabólicos complejos como el síndrome de Vici. Estos nuevos defectos, todos identificados por técnicas de secuenciación masiva sin marcadores metabólicos obvios, plantean la cuestión de una definición más amplia de LSD con la acumulación de material no digerible en el sistema endosómico/lisosómico.
4.3.3. Trastornos del ciclo vesicular sináptico: la sinapsis es una unión celular altamente especializada que conecta una neurona transmisora presináptica con una neurona receptora postsináptica. El concepto de “metabolismo sináptico” se ha introducido recientemente y podría definirse como la composición química específica y las funciones metabólicas que se producen en la sinapsis. La vesícula sináptica (SV) es un orgánulo complejo e independiente especializado. Las mutaciones que codifican muchas proteínas diferentes que regulan la vía exocítica-endocítica SV se han descrito como responsables de una variedad de trastornos que incluyen encefalopatías del desarrollo y parkinsonismos precoces, entre otros (18).
4.3.4. Deficiencias de aminoacil-ARNt sintetasas: con la excepción de GARS y KARS, los aaRS mitocondriales y citoplasmáticos están codificados por diversos genes nucleares. Las deficiencias de sintetasas de aminoacil-ARNt se presentan con un amplio espectro clínico con muchos fenotipos neurológicos. Las deficiencias de aaRS mitocondriales pueden simular trastornos de OXPHOS con hiperlactatemia. El diagnóstico debe facilitarse mediante ensayos de aminoacilación como se mostró recientemente para pacientes con mutaciones LARS2 y KARS. Las mutaciones en KARS y GARS, que actúan tanto en las mitocondrias como en el citosol, así como en varias sintetasas de aminoacil-ARNt citosólicas abren un campo nuevo en los EIM, ya que las sintetasas de ARNt citoplasmáticas son necesarias para la síntesis de todas las proteínas de la célula y, por lo tanto, para todos los orgánulos.
5.- GRUPO 3: TRASTORNOS RELACIONADOS PRINCIPALMENTE CON EL METABOLISMO ENERGÉTICO
Estos son EIM con síntomas debidos, al menos en parte, a una deficiencia en la producción o utilización de energía en cerebro, músculo, miocadio, hígado y otros tejidos. El diagnóstico puede orientarse mediante pruebas funcionales que miden glucosa, lactato, cetonas y otras moléculas energéticas (AA, ácidos orgánicos, acilcarnitinas) en sangre, LCR y orina y confirmadas por ensayos enzimáticos y pruebas moleculares. Los procesos de generación de energía son reguladores maestros de la vida celular. En particular, la mitocondria es tanto el origen como el objetivo de varias señales metabólicas que orquestan la función celular y la homeostasis. Las enfermedades mitocondriales son el grupo más común de EIM y se encuentran entre las formas más frecuentes de trastornos neurológicos hereditarios. La medicina mitocondrial se ha desarrollado en gran medida hasta convertirse en una subespecialidad bastante autónoma dentro de los EIM. Debido a su complejidad, destacaremos brevemente los subgrupos básicos y los conceptos centrales relacionados con el metabolismo energético.
5.1. Transportadores de membrana de moléculas energéticas
La glucosa, el lactato, piruvato y los cuerpos cetónicosson transportados mediante las proteínas codificadas por la familia de genes SLC, SLC2 y SLC5 codifican portadores de glucosa, GLUT y SGLT, respectivamente; mientras que la familia de genes SLC16 codifica para transportadores de monocarboxilados (MCT): AG, cuerpos cetónicos, lactato y piruvato. GLUT y MCT muestran muchas isoenzimas específicas de tejido como GLUT1 (el transportador cerebral de glucosa), GLUT2 (el transportador hepato-intestinal de glucosa) o MCT1.
5.2. Defectos de energía citoplasmática
Son, generalmente, menos severos. Incluyen EIM de la glucolisis, del metabolismo del glucógeno, de la gluconeogénesis e hiperinsulinismos que son tratables; trastornos del metabolismo de la creatina que son parcialmente tratables, y errores innatos de las vías de fosfato de pentosa, no tratables, con un fenotipo principalmente relacionado con la producción defectuosa de NADP/NADPH (19). El diagnóstico es habitualmente fácil de sospechar sobre una base clínica, además de las pruebas funcionales, la relación creatina/creatinina en orina y espectroscopía cerebral para defectos de creatina.
5.3. Defectos mitocondriales
Incluyen defectos de oxidación de la glucosa aeróbicos que se presentan con acidemia láctica congénita (transportador de piruvato, piruvato carboxilasa, piruvato deshidrogenasa y defectos del ciclo de Krebs), trastornos de la cadena respiratoria mitocondrial, transportadores mitocondriales de moléculas energéticas y otras moléculas indispensables, biosíntesis de coenzima Q, oxidación de AG y defectos de los cuerpos cetónicos. Algunos de los defectos de tiamina y riboflavina también pueden incluirse en este grupo dado que estas vitaminas sirven como cofactores para la descarboxilación oxidativa y la β-oxidación de ácidos grasos, respectivamente. Un grupo grande y creciente de trastornos (>110) involucra la maquinaria mitocondrial (fusión mitocondrial, fisión, replicación, importación de proteínas mitocondriales y control de calidad de proteínas, ribosomopatías, regulación del ADN mitocondrial y modificación intergenómica, sintetasas de ARNt mitocondriales y modificación de ARNt, y metabolismo de los fosfolípidos de membrana) (20). Las enfermedades mitocondriales son clínicamente diversas y pueden presentarse a cualquier edad. Pueden manifestarse de una manera específica de tejido o multisistémica, pero con mayor frecuencia afectan los órganos con las mayores demandas de energía, como el cerebro, el músculo esquelético, los ojos y el corazón. Alrededor de 290 genes registrados hasta ahora se clasifican en cinco categorías generales según Frazier y cols. (20). Las recomendaciones para un diagnóstico y tratamiento óptimo de este gran grupo en evolución se reevalúan constantemente.
6.- ASPECTOS RELEVANTES A TENER EN CUENTA
- Los EIM han experimentado una evolución muy importante en los últimos años en relación al número y complejidad de los mismos, lo cual ha modificado la definición intrínseca de lo que la comunidad científica considera como enfermedad metabólica hereditaria.
- Clasificar los EIM en función de su fisiopatología y ligarlo con las manifestaciones clínicas corresponde a una necesidad de entender, aclarar y comprender un campo muy diverso y difícil en cuanto a mecanismos y bases bioquímicas y biológicas. Además, pretende ser una herramienta práctica para el clínico.
- Las clasificaciones, o intentos de ellas, son propuestas temporales que han de ir renovándose constantemente en función de los conocimientos y avances de cada momento.
- La división en los tres grupos propuestos en la clasificación simplificada, con sus correspondientes subgrupos, no impide que un mismo EIM pueda pertenecer a varios de estos subgrupos, puesto que los mecanismos de enfermedad son diversos y múltiples.
BIBLIOGRAFÍA
- Morava E, Rahman S, Peters V, Baumgartner MR, Patterson M, Zschocke J. Quo vadis: the re-definition of “inborn metabolic diseases”. J Inherit Metab Dis. 2015; 38: 1003-6.
- Lamari F, Mochel F, Saudubray JM. An overview of inborn errors of complex lipid biosynthesis and remodeling. J Inherit Metab Dis. 2015; 38: 3-18.
- García-Cazorla A, Dionisi-Vici C, Saudubray JM. Disorders of cellular trafficking. In: Inherited metabolic disorders. Diagnosis and treatment. 7th ed. Springer; 2021.
- Ferreira CR, van Karnebeek CDM, Vockley J, Blau NA. Proposed nosology of inborn errors of metabolism. Genet Med. 2019; 21: 102-6.
- Saudubray JM, Mochel F, Lamari F, García-Cazorla A. Proposal for a simplified classification of IMD based on a pathophysiological approach: A practical guide for clinicians. J Inherit Metab Dis. 2019; 42: 706-27.
- Ferreira C, Rahman S, Keller M, Zschocke J, ICIMD Advisory Group. An International Classification of Inherited Metabolic Disorders (ICIMD). J Inherit Metab Dis. 2021; 44(1): 164-77.
- Tuschl A, Clayton PT, Gospe SM, Gulab S, Ibrahim S, Singhhi P, et al. Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man. Am J Hum Genet. 2012; 90: 457-66.
- Kremer LS, Danhauser K, Herebian H, Pedkovic Ramadza D, Piekutowska-Abramczuk D, Seibt A, et al. NAXE Mutations disrupt the cellular NAD(P)HX repair system and cause a lethal neurometabolic disorder of early childhood. Am J Hum Genet. 2016; 99: 894-902.
- Tărlungeanu DC, Deliu E, Dotter CP, Kara M, Janiesch PC, Scalise M, et al. Impaired amino acid transport at the blood brain Barrier is a cause of autism spectrum disorder. Cell. 2016; 167: 1481-94.
- Guemez-Gamboa A, Nguyen LN, Yang H, Zaki MS, Kara M, Ben-Omran T, et al. Inactivating mutations in MFSD2A, required for omega-3 fatty acid transport in brain, cause a lethal microcephaly syndrome. Nat Genet. 2015; 47: 809-13.
- Acuna-Hidalgo R, Schanze D, Kariminejad A, Nordgren A, Hasan Kariminejad M, Conner P, et al. Neu Laxova syndrome is a heterogeneous metabolic disorder caused by defects in enzymes of the L-serine biosynthesis pathway. Am J Hum Genet. 2014; 95: 285-93.
- Boycott KM, Beaulieu CL, Kernohan KD, Gebril OH, Mhanni A, Chudley AE, et al. Autosomal recessive intellectual disability with cerebellar atrophy syndrome caused by mutation of the manganese and zinc transporter gene SLC39A8. Am J Hum Genet. 2015; 97: 886-93.
- Dubail J, Huber C, Chantepie S, Sonntag S, Tüysüz B, Mihci E, et al. SLC10A7 mutations cause a skeletal dysplasia with amelogenesis imperfecta mediated by GAG biosynthesis defects. Nat Commun. 2018; 9: 3087.
- Vianey-Saban C, Acquaviva C, Cheillan D, Collardeau-Frachon S, Guibaud L, Pagan C, et al. Antenatal manifestations of inborn errors of metabolism: biological diagnosis. J Inherit Metab Dis. 2016; 39: 611-24.
- Ferdinandusse S, Falkenberg KD, Koster J, Mooyer PA, Jones R, van Roermund CWT, et al. ACBD5 deficiency causes a defect in peroxisomal very long-chain fatty acid metabolism. J Med Genet. 2017; 54: 330-7.
- Coman D, Vissers LELM, Riley LG, Kwint MP, Hauck R, Koster J, et al. Squalene synthase deficiency: Clinical, biochemical, and molecular characterization of a defect in cholesterol biosynthesis. Am J Hum Genet. 2018; 103: 125-30.
- Mills E, Green R. Ribosomopathies: There’s strength in numbers. Science. 2017; 358: 608-16.
- Cortès-Saladelafont E, Lipstein N, García-Cazorla A. Presynaptic disorders: a clinical and pathophysiological approach focused on the synaptic vesicle. J Inherit Metab. 2018; 41: 1131-45.
- Qian Y, Banerjee S, Grossman CE, Amidon W, Nagy G, Barcza M, et al. Transaldolase deficiency influences the pentose phosphate pathway, mitochondrial homeostasis, and apoptosis signal processing. Biochem J. 2008; 415: 123-34.
- Frazier AE, Thorburn DR, Compton AG. Mitochondrial energy generation disorders: genes, mechanisms and clues to pathology. J Biol Chem. 2019; 294(14): 5386-95.